
Curious about the brain
driven to make a difference
Pioneering discoveries in neuronal cell death, survival, and the molecular mechanisms of neurodegenerative diseases.

The BRAIN Lab
Biology of Regulated cell death and Advanced Investigation in Neurodegeneration
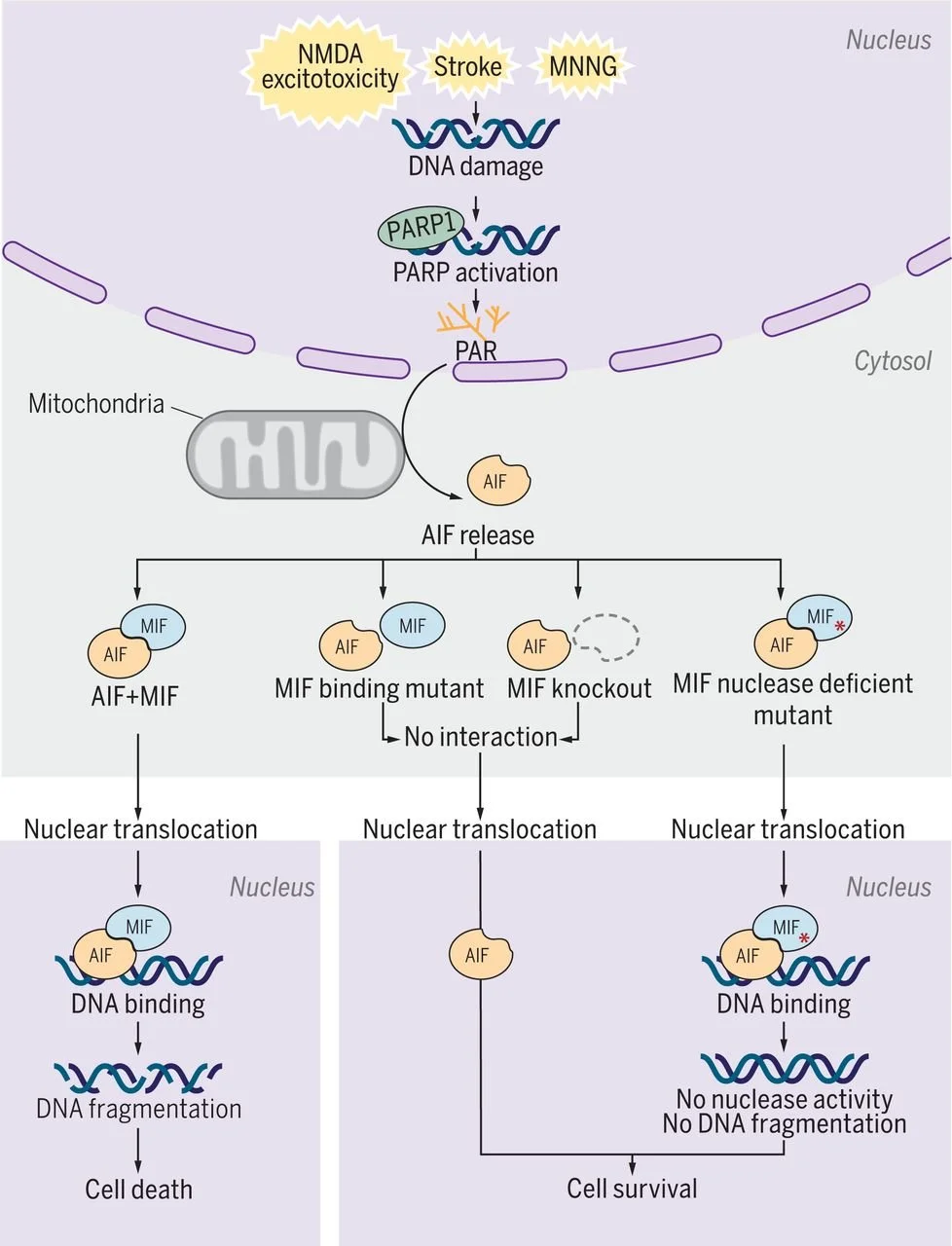
The BRAIN Lab brings together seven labs to uncover how brain cells die and survive, driving discoveries in Parkinson’s, Alzheimer’s, and stroke. We pioneered Parthanatos and identified key disease genes, with therapies now advancing toward the clinic.
Our mission is simple yet ambitious: to transform fundamental discoveries into treatments that slow or stop disease progression and improve patients’ lives.
UNRAVELING THE MYSTERIES OF NEURODEGENERATION





LEADERSHIP
Ted M. Dawson, MD, PhD
Co-Director, The BRAIN Lab
Director, Institute for Cell Engineering
Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases
Professor of Neurology, Neuroscience, Pharmacology, and Molecular Sciences


Valina L. Dawson, PhD
Co-Director, The BRAIN Lab
Director, Neuroregeneration and Stem Cell Programs, Institute for Cell Engineering
Professor of Neurology, Neuroscience, and Physiology






COLLABORATING LABS
-

THE DAWSON LAB
-

The Kam Lab
-

The Kang Lab
-

The Sachdeva Lab
-

The Xu Lab
-

The Mao Lab
-

The Ko Lab